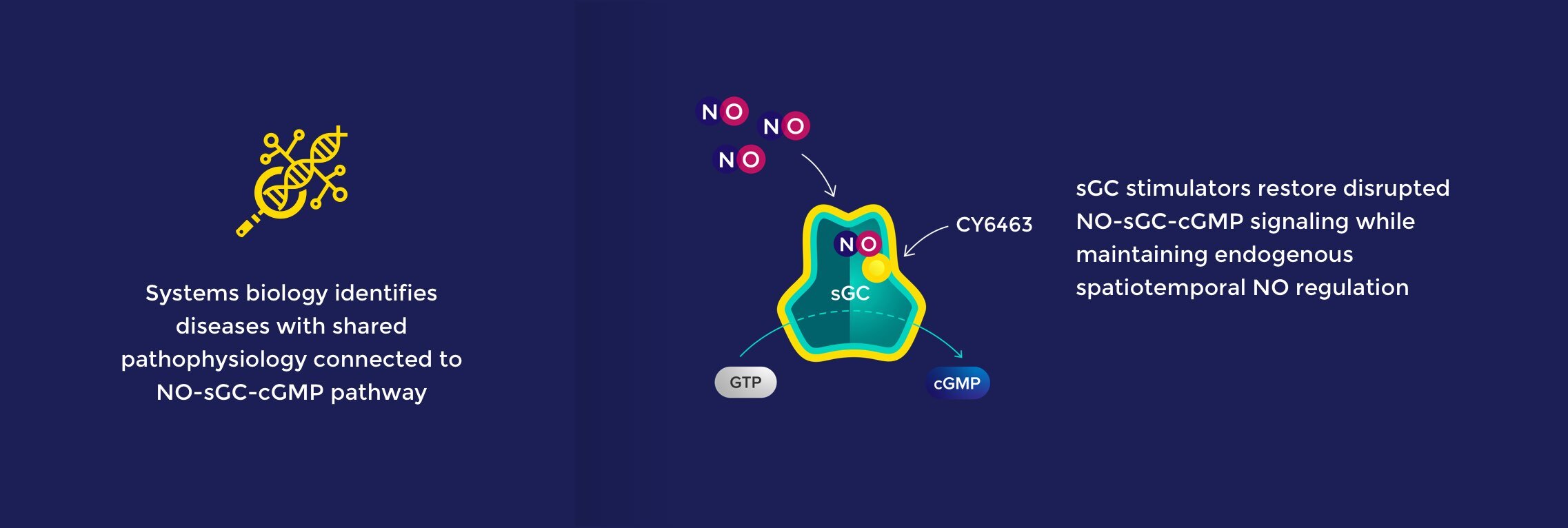
NO-sGC-cGMP Pathway
The nitric oxide (NO) – soluble guanylate cyclase (sGC) – cyclic guanosine monophosphate (cGMP) signaling pathway is a fundamental mechanism that precisely controls key aspects of physiology throughout the body. The NO-sGC-cGMP pathway regulates diverse and critical biological functions and has been successfully targeted with medicines approved for cardiovascular and pulmonary indications.
Praliciguat is a systemic sGC stimulator that is licensed to Akebia Therapeutics, Inc. and being advanced in rare kidney disease.
Olinciguat is a clinical-stage vascular sGC stimulator that we intend to out-license for cardiovascular diseases.
Zagociguat is a clinical-stage CNS-penetrant sGC stimulator that has shown rapid improvement in cerebral blood flow, functional brain connectivity, brain response to visual stimulus, cognitive performance, and biomarkers associated mitochondrial function and inflammation in clinical studies.
CY3018 is a CNS-targeted sGC stimulator that preferentially localizes to the brain and has a pharmacology profile that suggests its potential for the treatment of neuropsychiatric diseases and disorders. Zagociguat and CY3018 were sold to Tisento Therapeutics, Inc., a company in which Cyclerion holds a 10% equity stake.
Cyclerion is actively evaluating other activities aimed at enhancing shareholder value, which may potentially include collaborations, licenses, mergers, acquisitions and/or other targeted investments.